Products
Applications
Learning
 Part of the Oxford Instruments Group
Part of the Oxford Instruments Group
30th March 2023 | Author: Robin Blagg
I joined Oxford Instruments Magnetic Resonance in the autumn of 2020, after spending over a dozen years performing postgraduate and postdoctoral research, at various UK universities. During this time I’d made significant use of high-field NMR spectroscopy for such tasks as structural elucidation, reaction monitoring, and analysis of dynamic processes in solution; however I’d never used a benchtop NMR spectrometer.
Over time, as I become more experienced with our X-Pulse Broadband Benchtop NMR Spectrometer, I realised that not only could a significant proportion of all the NMR spectroscopy I’ve performed over the years have been performed on the X-Pulse. But also, how access to an X-Pulse would have allowed for more experiments to be performed making me more productive than was otherwise possible.
In this blog, I therefore discuss the ways I used high-field NMR spectroscopy during my research career, and how I could have made use of benchtop NMR spectroscopy for the same applications.
NMR spectroscopy is arguably one of the most powerful tools in the synthetic chemists toolbox, for the characterisation of a chemical compound or analysis of a mixture. As an organometallic chemist I benefitted from NMR spectrometers able to observe a wide range of nuclei; not only those stalwarts of organic chemistry 1H & 13C; but also, depending on the chemistry of interest, 19F, 11B & 31P. Whilst the spectrometers I used, ranged in frequency between 300 & 500 MHz (many of which used Oxford Instruments superconducting magnets).
An example of the sort of compounds I characterised using NMR spectroscopy are a series of rhodaboratrane complexes I synthesised during my PhD at the University of Bristol; these were characterised by 1H, 11B{1H}, 13C{1H} and 31P{1H} NMR spectroscopy (along with other techniques), representative 1H & 11B{1H} spectra are shown in figure 1. The 11B{1H} spectrum (figure 1, right) shows two signals; the major component giving a doublet at δB +9 ppm, arising from J-coupling between boron and rhodium; while the minor component (a known by-product of the reaction) gives a single peak at δB +1 ppm. In the 1H NMR spectrum (figure 1, left) the two chemical environments of the thioxotriazole bridging heterocycle (sulfur trans or cis to CO) can clearly be observed around δH +2.3 ppm in a 6:3 ratio (the same can be seen for the ethyl CH2 quartets around δH +3.8 ppm).
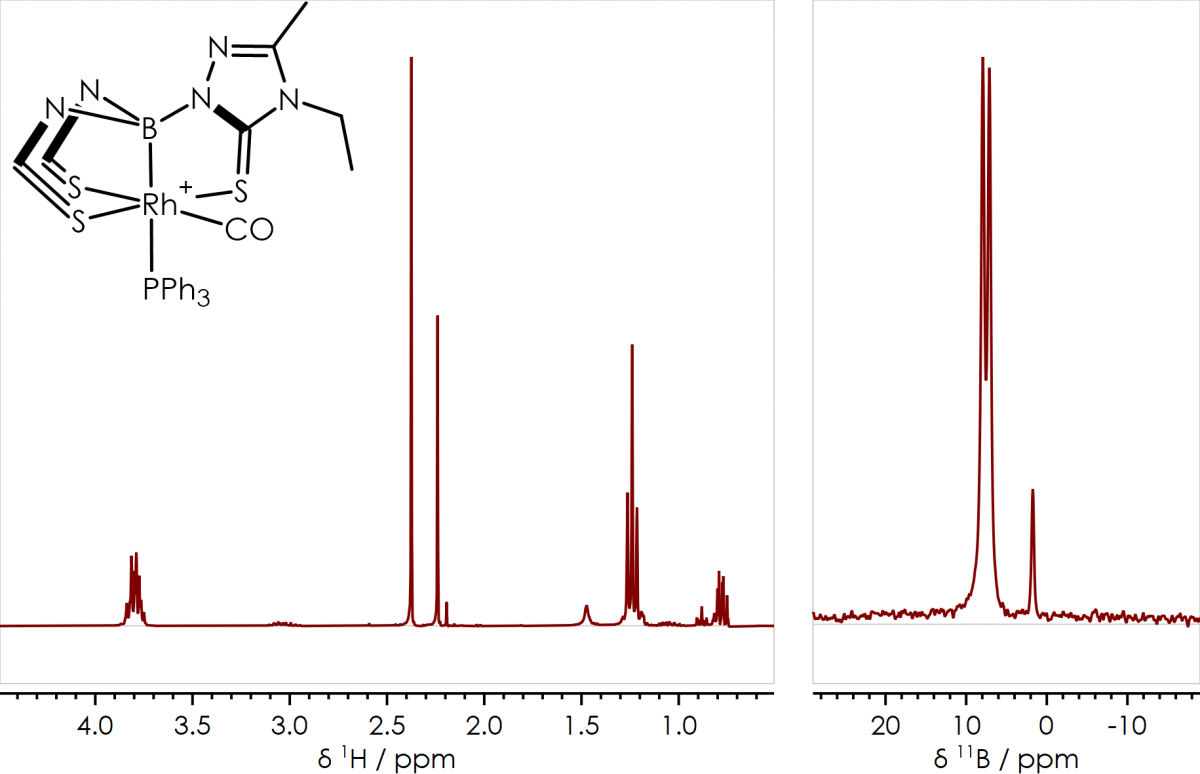
Figure 1 [Rh(CO)(PPh3){B(taz)3}]+ 1H & 11B{1H} NMR Spectra & Structure, acquired on a 300 MHz instrument [Dalton Trans., 2009, 8724-8736]
While these spectra were acquired using a 300 MHz superconducting magnet, there’s no reason I couldn’t have obtained the same information from a 60 MHz permanent magnet based system with broadband capability, such as the X-Pulse.
There’s one compound I’ve previously synthesised, where NMR spectra obtained at various frequencies can be compared. I made significant use over the years of tetrabutylammonium tetrakis(pentafulorophenyl)borane, [Bu4N][B(C6F5)4], a non-coordinating electrolyte. Following synthesis 1H, 11B & 19F NMR spectra would be used to confirm purity; and the same information can as readily be obtained at benchtop as could be obtained on high-field systems (figure 2). Indeed, a signal arising from a trace impurity can readily be observed (δF −165.5 ppm) in the sample analysed on the X-Pulse (from a batch I did not synthesise), while no impurities can be observed in the sample analysed on a high-field system (from a batch I did synthesise).
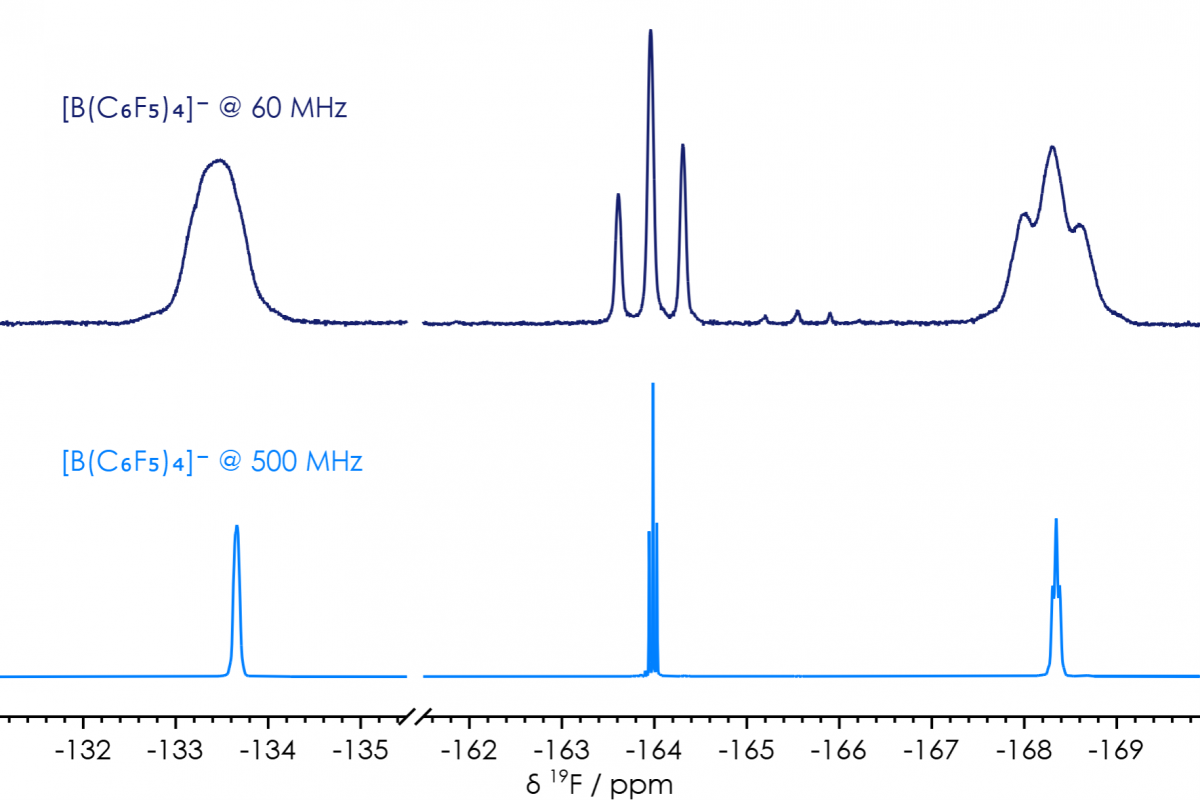
Figure 2 [B(C6F5)4]− 19F NMR Spectra, comparison of high-field & benchtop
This compound is also an example where not everything observed at high-field can readily be observed on benchtop. Specifically, the signals in the carbon-13 NMR spectrum, associated with the C6F5 rings. While in the spectrum collected in ca 25 min on a 500 MHz high-field system, the signals can be observed in the range δC +155 - +120 ppm, with signal-to-noise ratios (SNRs) around six (compared with SNR >100, for the sharp signals from the butyl groups). While, for a comparable sample on a 60 MHz benchtop system, even after a ca 24 hour acquisition, these signals can only be inferred by slight imperfections in the baseline (figure 3).

Figure 3 [Bu4N][B(C6F5)4] in CD3CN, 13C{1H} NMR Spectra, comparison of high-field & benchtop
One other difference I found when moving to benchtop, was the difference in priority for some of the more advanced, two-dimensional homo- and particularly hetero-nuclear correlation experiments. While at high-field a carbon-13 spectrum can often be obtained in under half an hour, an equivalent measurement can often take multiple hours for all but the highest sample concentrations at benchtop. Meanwhile, a 1H-13C correlation experiment will take a couple of hours at both high-field and benchtop (since the total acquisition time is more dependent on the number of slices necessary to build the second dimension), therefore it’s often more time efficient (and informative) to obtain gradient-selective two-dimensional spectra like HSQC & HMBC than a simple one-dimensional 13C spectrum when operating at benchtop frequencies.
In general, with exception of some of the 13C NMR spectra, all of the structural elucidation I performed on high-field systems over the years, could have been performed on the X-Pulse; in my case usually still acquiring just simple one-dimensional NMR spectra, though with the option to obtain advances one- and two-dimensional spectra as required.
Since starting to work with the X-Pulse, not only have I found that a significant proportion of the NMR spectroscopy I’ve previously performed on high-field systems could have been performed on the X-Pulse. But the convenience factor of having day-to-day access to a benchtop system in the lab, would actually have allowed me to be more productive, by performing more measurements that just wouldn’t have been convenient to perform on communal high-field systems.
Robin Blagg,
Applications Scientist, Oxford Instruments
Robin Blagg is the applications team leader at Oxford Instruments Magnetic Resonance. Robin completed his first degree at the University of York, then obtained his PhD in organometallic chemistry at the University of Bristol, and then undertook post-doctoral research at the Universities of Sussex, Manchester, and East Anglia; before joining Oxford Instruments in October 2020. Robin is a member of the Royal Society of Chemistry (RSC), and serves on the committee of the RSC NMR Discussion Group.

© Oxford Instruments 2025